At the moment, this is a brief and selective history of the theory of gas-surface dynamics. I may include some on experimental work, but then who would believe that I know any of that ?
The really key early work is undoubtedly that of Lennard-Jones. In his now classic treatise Processes of Adsorption and Diffusion on Solid Surfaces, he invented a description simple enough and yet sufficiently fundamental that we still use it today.
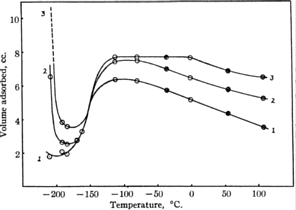
But first, you have to know what he was trying to explain. In the 1920s, some experiments were performed at Princeton to measure the uptake (volume adsorbed, i.e. amount that sticks to the surface) of a gas in equilibrium with the surface. The gas in question being hydrogen, which is a popular gas to work with, as you will discover if you read much more of this guide.
These experiments were not like modern experiments; they did not measure the probability of sticking to a surface as a function of molecular energy. The apparatus was made of glass (19k GIF), it was not a massive stainless steel thing!, and the main variable controlled was the temperature in the reaction flask, i.e. the temperature of both the gas and the surface.
The sort of results obtained when the surface was Ni are shown here.
So what does this mean ?
This was called abnormal adsorption. What they thought of
as normal was that the uptake should simply decrease as the temperature
increases. The idea was that if you increase the temperature, the gas molecules
and surface atoms all move faster, so it becomes harder and harder for them to
stick together after a collision - they're moving so fast they just bounce off.
In this case, it was suggested that there is a reaction, which is in some
way activated. The low temperature behaviour was described as a
secondary process corresponding to reversible adsorption of intact
H2 molecules amenable to classical interpretation, while the high
temperature data was called primary adsorption, an activated process
which may even correspond to complete dissociation.
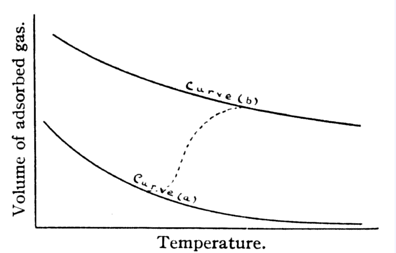
So the model is one of two states, ie two processes: Both of these processes will have adsorption isobars (i.e. graphs like the
one above) which simply decay with temperature. However, if (a) has a smaller
heat of adsorption than (b) (i.e. you get less energy released when (a) happens
than when (b) happens) and there's a bigger energy barrier for (b) to happen,
then the isotherm for (b) will lie above that for (a). At low temperature you
can imagine that there is only enough energy for (a) to happen, but as you make
it hotter, eventually (b) takes over. This is shown schematically below.
(a) S + H2 -> S(H2), i.e. the adsorbed molecule
(b) S + H2 -> S(2H), i.e. adsorbed atoms from the dissociated
molecule.
So there are two ways of adsorbing, as an intact molecule and as separated atoms, i.e. dissociative adsorption. What Lennard-Jones did was to link these vague ideas with energetics: the Lennard-Jones model is a model for how the energy of the molecule (intact or dissociated) changes with distance from the metal surface.
The molecule-metal interaction is represented by curve (1). At
some distance from the surface there is a minimum in the potential energy
arising from competition between the van der Waals attraction and the strong
repulsion due to the overlap between molecular and metal electron clouds.
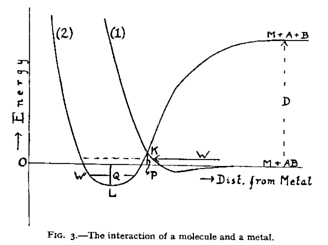
The molecule can be dissociated far from the surface, by adding the (large) amount of energy D. Bonding/anti-bonding interactions mean that these atoms are then very strongly attracted to the surface. In equilibrium they will be located at the minimum, L.
A dissociation event, like (b) above, occurs if an intact molecule approaches the surface until K, where it makes a radiationless transition from (1) to (2) becoming adsorbed as atoms.
You may think What's the big deal?, but what Lennard-Jones did was to add to the picture some understanding of energetics. Also the states - intact molecule or two atoms - are associated with two diabatic states between which the molecule can make transitions. These can be re-expressed as adiabatic states and the debate is still ongoing today as to which description diabatic or adiabatic (with attendant, usual approximations) is best used in any particular circumstance.
Another point is that the Lennard-Jones model is easy to extend by adding
extra diabatic curves between (1) and (2), which could represent excited
molecular states or molecular ions. If these states have wells at different
distances from the surface, then there could be complicated trapping with a
strong influence on the dissociation. This is known as
Precursor Mediated Dissociation.
Although it is useful, there is an inconsistency in the model in that when changing from (1) to (2) the molecular bond is instantaneously elongated. Lennard-Jones noted that although one-dimensional potential energy curves can prove of great value in discussions, they do not lend themselves to generalisation when more than one coordinate is necessary to specify a configuration. In a quantitative theory, there should be a number of additional curves between (1) and (2) corresponding to rotational and vibrational states of the molecule. In fact adding vibrations leads to the now famous elbow potential, which appears in some form in almost all modern work.
To illustrate increasing dimensionality, the effects of including the surface periodicity was studied as an example.
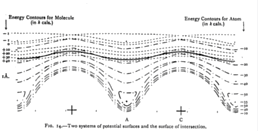 |
In this plot, the two previous curves, (1) and (2) have been extended in the direction across the surface and the curve crossing point becomes a seam of intersection along which it is energetically possible for the molecule to dissociate at no energy cost. |
More modern history
Some of the original source material: H. S. Taylor, Journal of the American Chemical Society 53,
(1931) 578.
A. F. Benton and T. A. White, Journal of the American Chemical Society
53 52, (1930) 2325.
J.E. Lennard-Jones, Transactions of the Faraday Society 28,
(1932) 333.
©G.R. Darling
Return to index.
Return to my homepage.
Comments to
darling@ssci.liv.ac.uk